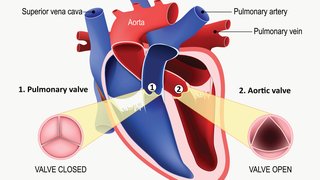
For nearly 150 years after its discovery, cardiac amyloidosis was considered essentially untreatable. Characterized by proteins that "misfold," preventing them from retaining their normal structure, amyloidosis causes a buildup of misfolded proteins that bog down the work of the heart and organs throughout the body –like too much mortar in a brick wall.
Historically, cardiac amyloidosis could only be diagnosed at medical centers like UT Southwestern that have the expertise to do endomyocardial biopsies, which involve removing and testing small samples of heart tissue. Then, add the complexity of disease typing – there are 36 known subtypes of amyloidosis, 11 of which are known to infiltrate the heart. Of these 11, 95% are diagnosed as either amyloid light chain (AL), which is caused by abnormal bone marrow cells, or transthyretin amyloidosis (ATTR), which is caused by deposits in the heart of the blood protein transthyretin (TTR).
The inherent difficulty of diagnosing and managing cardiac amyloidosis contributed to a culture of apathy toward the condition among many providers. “Why search for a complex disease…” the rationale went, “when there is no cure, let alone an effective treatment?”
But in the last decade, there has been significant progress on three fronts of cardiac amyloidosis management:
- Less invasive, more affordable diagnosis: Endomyocardial biopsy is no longer the only way to diagnose ATTR-CA. Advances in diagnostic technology, including imaging, blood, and urine tests, can be used to confirm ATTR in cases in which we can rule out AL amyloidosis.
- Effective treatments: In 2018 and 2019, the first treatments for ATTR were approved by the U.S. Food and Drug Administration (FDA). They have been shown to slow disease progression, reduce hospitalizations, and cut the risk of cardiac death. In parallel, advances in chemotherapy have transformed the prognosis for patients with AL by improving survival and, possibly, function of the affected organs.
- Research: Clinical studies, including several at UT Southwestern, are taking deeper dives into specific subtypes of cardiac amyloidosis in search of more evidence about genetic biomarkers and potential new medications.
UTSW clinicians and researchers have remained at the forefront of innovation in amyloidosis management. Our Multidisciplinary Amyloidosis Program is a recognized treatment center by the Amyloidosis Foundation, and UT Southwestern is a Rare Disease Center of Excellence.
Spotting AL or ATTR-CA amyloidosis early is the key to giving more patients the chance for longer, healthier lives. As knowledge and awareness of cardiac amyloidosis continues to grow, so does the need to recognize and investigate its vague yet telltale symptoms.
Building an index of suspicion for cardiac amyloidosis

Though cardiac amyloidosis typically develops over several years, it can progress rapidly to a severe illness. Research has shown that there are many cardiac and non-cardiac signs that should be investigated as early as possible.
For example, a study published in 2019 showed that about 10% of men older than 50 or women over 60 with carpal tunnel syndrome in both wrists had amyloid tissue – a potential precursor of cardiac or systemic amyloidosis. Other potential amyloidosis symptoms include:
- Fatigue
- Chest discomfort (angina)
- Heart palpitations
- Thick heart muscle in the left ventricle of the heart, which pumps blood out of the heart through the aorta
- Enlarged tongue
- Purple color around the eyes
- Chronic diarrhea or constipation
- Loss of bladder control
- Low blood pressure
- Spontaneous tendon rupture
- Swelling of the abdomen, legs, or ankles
- Neuropathy (lost sensation or pins and needles) in the arms, legs, or feet that isn’t associated with diabetes or another known cause
- Narrowing of the spine (spinal stenosis)
- Family history of amyloidosis, as some subtypes of ATTR can be inherited
The most critical step in building our index of suspicion for cardiac amyloidosis is to rule out AL amyloidosis. That is because the average lifespan after diagnosis is just 6 months without treatment, compared to ATTR-CA, which carries a lifespan of 4-6 six years without treatment.
Diagnosing AL and ATTR cardiac amyloidosis
If AL is identified or can’t be ruled out, the diagnostic pathway looks very different from the steps to diagnose ATTR-CA. In 2023, the American College of Cardiology provided the latest expert guidance on clinical diagnosis and treatment pathways for AL and ATTR amyloidosis.

Looking for abnormal light chains
Suspecting or spotting AL amyloidosis through a blood test signals the start of a diagnostic emergency – establishing a tissue-based diagnosis is crucial. In many cases, we can rule out AL amyloidosis with a blood test that looks for abnormal light chains in the patient’s blood work.
Light chains are proteins that are made by the plasma cells are supposed to either bind to heavy chains to form immunoglobulins to fight illnesses or circulate in the blood. When too many light chains are circulating, it can signal AL amyloidosis.
Patients with a high volume of light chains should be evaluated within a few days with a bone marrow histopathology workup and endomyocardial biopsy. An EKG can detect an unusual “low-voltage pattern,” and an echocardiogram can gauge whether the left and right ventricles are thicker than normal. Together, these tests can establish very strong evidence of cardiac amyloidosis.
By expediting these diagnostic tests, we can start treatment as quickly as possible for AL amyloidosis – or proceed with diagnostic testing for ATTR.
Nuclear medicine scan, virtual biopsy for ATTR
While we still want to move quickly, diagnosing ATTR is less urgent compared with AL amyloidosis. More than 10% of patients are symptomatic for three years or longer before being diagnosed with cardiac ATTR-CA.
When we can’t rule out AL, an endomyocardial biopsy is needed to diagnose suspected ATTR-CA or AL due to the presence of an abnormal protein in the blood called monoclonal gammopathy of uncertain significance (MGUS). Having MGUS doesn’t cause symptoms, but it can indicate another condition such as AL amyloidosis or multiple myeloma, a type of blood cancer.
If AL amyloidosis is ruled out, we can do noninvasive diagnostic testing for cardiac ATTR:
- Scintigraphy, which is a new implementation of an existing nuclear medicine scan that uses a special bone radiotracer called technetium pyrophosphate (99mTc-PYP) to help us distinguish various forms of amyloids.
- Blood and urine samples test for abnormal monoclonal proteins that may indicate the presence of AL amyloidosis – another step to rule out that form of amyloidosis.
Another test that is commonly ordered is a cardiac MRI, which serves as a form of a virtual biopsy. Using a contrast agent called gadolinium, we can see with exquisite detail what is going on within the heart, along with its natural contours, size, shape, and any amyloidosis fibrils inside. Whereas cardiac MRI can’t tell us the type of amyloidosis, it can help distinguish between amyloidosis and other types of heart disease.
Treating cardiac amyloidosis
Coordinated care and early treatment is essential to achieve the best outcomes for cardiac amyloidosis. Our Multidisciplinary Amyloidosis Program includes specialized nurses and experts from hematology and oncology, myeloma specialists for AL, cardiology, cardiac imaging, genetics, gastroenterology, and neurology to meet each patient’s needs.
Advanced therapies for AL
Our patients can get the highly effective combination of two advanced therapies:
- CyBorD, a chemotherapy combination that includes cyclophosphamide, bortezomib, and dexamethasone
- Daratumumab, a monoclonal antibody drug, which was recently recognized for its ability to slow the progression of AL amyloidosis
The phase-3 ANDROMEDA study showed that this combination provided a 96% hematologic response in patients with a median of two involved organs, such as the heart and kidneys. More than half of patients (54%) had a complete response with 15 months of infusions.
FDA-approved drugs for ATTR
The last five years have been transformative in ATTR treatment. In 2018 and 2019, new treatments were developed that stabilize the TTR protein or silence the translation of the TTR mRNA into protein. Also in 2018, inotersen and patisiran were FDA-approved for hereditary ATTR that causes neuropathy. UT Southwestern was among the first sites in Dallas to give a patient a patisiran infusion.
In 2019, tafamidis became the first FDA-approved drug for treating hereditary and wild-type ATTR that causes cardiomyopathy. It acts like a bit of glue to hold the protein folds in place, stabilizing them so they don't misfold. Prior to its FDA approval, UT Southwestern patients had access to tafamidis through clinical trials. Tafamidis doesn't cure or reverse ATTR, but it has been shown to:
- Slow disease progression
- Reduce the frequency of heart-related hospitalizations
- Reduce the risk of cardiac death by 30%
Related reading: New drugs for cardiac amyloidosis provide hope for patients
Research in cardiac amyloidosis is thriving
After decades of limited research outside of amyloidosis centers, new clinical studies are surfacing around the U.S., including at UT Southwestern.
In August 2022, we launched a clinical trial focused on the genetic mutation that is the most common mutation responsible for hereditary ATTR-CA. About 2% to 4% of African Americans and people of Afro-Caribbean descent carry this mutation, making them more vulnerable to developing amyloidosis. We are enrolling patients across other sites in the U.S. to gain a better understanding of specific clues we can use to identify carriers early and intervene before the disease becomes severe.
A few more research highlights include:
- CAEL-101 monoclonal antibody study: UT Southwestern is enrolling patients in a clinical trial to determine whether this treatment is well-tolerated and can improve survival in patients with advanced AL amyloidosis (stage IIIb). This study is offered at the Harold C. Simmons Comprehensive Cancer Center.
- Cardio TTRANSFORM: This clinical trial is currently recruiting patients to evaluate the effectiveness of eplontersen, a ligand-conjugated antisense medication designed to relieve and prevent symptoms caused by hereditary transthyretin-mediated amyloid polyneuropathy. The medication silences the translation of TTR mRNA into protein.
- Safety, Efficacy, and Pharmacokinetics of Tafamidis: This study in patients with transthyretin-mediatric amyloidosis post-orthotopic heart transplantation is not yet recruiting. It will be a single-arm intervention clinical trial to gather observations to establish the role of tafamidis use for the management of ATTR in patients post-heart transplant.

Join our research efforts
In the future, we hope to become a site for a trial of CRISPR CAS9, a gene-editing technology. At the AHA Scientific Sessions in November 2022, UK researchers shared results from their phase 1 clinical trial, which investigated the tolerability and effectiveness of two dosage levels of CRISPR CAS9 medication NTLA-2001 in patients with hereditary ATTR with polyneuropathy and patients with ATTR cardiomyopathy. Within 28 days, both dosage levels reduced circulating TTR protein in these patients by 90%, which could lead to better cardiac outcomes.
Parallel advances in diagnosis, treatment, and research of cardiac amyloidosis have given new hope to patients with this previously untreatable disease – and new pathways for providers to identify and intervene earlier, when the condition is potentially more treatable.
If you or your doctor suspect you have cardiac amyloidosis, visit with a specialist as soon as possible. With advanced treatment and coordinated care, more patients are living longer, healthier lives.
To talk with a cardiac amyloidosis specialist, call 214-645-8300 or request an appointment online.